



It began with a simple question: why do some patients with beta-thalassaemia live relatively normal lives, while others depend on lifelong blood transfusions?
For Malaysian-born consultant haematologist Dr Thein Swee Lay, that question would spark a decades-long scientific journey.
Her research uncovered a hidden genetic “switch” and paved the way for breakthrough treatments for the inherited blood disorders beta-thalassaemia and sickle cell disease.
Born and raised in Malaysia, Dr Thein completed her medical degree at Universiti Malaya before moving to Britain for postgraduate training.
“I was drawn to haematology because it allowed me to combine laboratory-based research with clinical work, while continuing to see patients,” she shares.
After completing her training, she joined the University of Oxford’s Medical Research Council (MRC) Molecular Haematology Unit under the late physician and molecular genetics researcher Sir Prof Dr David Weatherall.
This proved to be a pivotal moment that marked the start of her research career.
“It began with my mentor, Sir David, posing a very simple scientific question: whether I could investigate why some patients with beta-thalassaemia have a much milder form of the disease, unlike the majority who depend on lifelong blood transfusions.
“This led me to understand how complex genetics can be, and to unravel why patients with the same mutation can experience vastly different disease severity,” she says.
“It appears to be related to other genes inherited alongside the beta-thalassaemia gene.”
Tracking genetic patterns
To find answers, she turned to patients themselves, eventually following genetic clues across continents.
First, Dr Thein began recruiting patients and their families who had unusually mild beta-thalassaemia.
This took her across Britain and beyond, including to the Italian island of Sardinia (one of the world’s Blue Zones) and the African country of Malawi.
One key pattern soon emerged.
“Many of these patients had milder disease because they continued producing high levels of fetal haemoglobin, which compensated for their inability to produce adult haemoglobin,” she explains.
Another important observation was that this ability was inherited independently of the beta-globin gene – the gene where the disease-causing mutation occurs.
At the time, technology was far less advanced, and identifying such genes required studying large families to strengthen the findings.
Dr Thein used linkage analysis to track genetic patterns across generations.
“I was fortunate to identify one such family, some of whom lived in Malawi,” she says.
This led to the identification of a gene on chromosome 6q, separate from the beta-globin gene on chromosome 11.
“However, this gene did not explain the high fetal haemoglobin levels in other families, suggesting that more than one gene was involved.”
From roadblock to breakthrough
With her findings unable to explain all cases, Dr Thein’s research spluttered to a stop.
“At that point, I hit a roadblock because the other families were smaller and lacked the statistical power to identify additional genes,” she says.
“So I turned to twin studies.”
Twin studies are a powerful way to determine whether a trait has a genetic influence.
She collected data from more than 5,000 twins, comparing fetal haemoglobin levels between identical and non-identical pairs.
The results showed that identical twins had much more similar levels, indicating a strong genetic component.
By then, genetic technology had advanced, and genome-wide association studies had become available, allowing researchers to perform an unbiased scan of the genome.
“This enabled us to identify genetic differences between individuals with very high and very low levels of fetal haemoglobin,” she says.
The approach led to the discovery of three genes.
One was on chromosome 11, where the beta-globin gene is located.
The second was on chromosome 6q, which Dr Thein had identified earlier.
The third was BCL11A – a gene not previously known to play any role in haemoglobin production.
“Once we discovered it, I was able to show that BCL11A was responsible for the high levels of fetal haemoglobin seen in some patients with beta-thalassaemia and sickle cell disease,” she says.
The ‘brake’ in our blood
But identifying BCL11A was only the beginning; understanding how it worked proved to be the real breakthrough.
“We later realised that BCL11A acts as a repressor – switching off the production of fetal haemoglobin as the body transitions to adult haemoglobin,” says Dr Thein.
In some individuals, however, this “brake” is lifted, allowing them to continue producing fetal haemoglobin.
“Fetal haemoglobin is something all of us produce while in the womb,” she explains.
“Even after birth, small amounts remain, but most haemoglobin switches to the adult form, which carries oxygen in the blood.”
While we’re in the womb, fetal haemoglobin is produced by the gamma-globin gene.
Just before birth, a “switch” occurs, and production shifts to adult haemoglobin, which is made by the beta-globin gene.
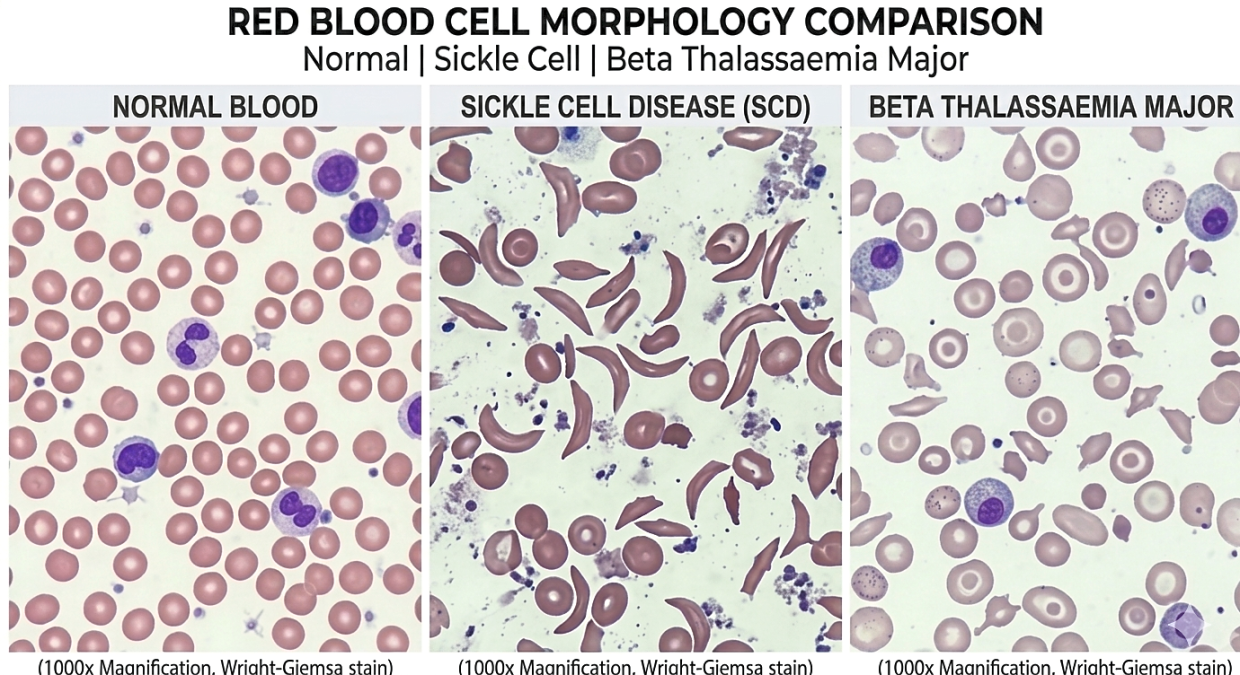
Says Dr Thein: “In conditions such as beta-thalassaemia and sickle cell disease, where the beta-globin gene is defective, this switch leads to complications.
“However, in patients with milder forms of the disease, the switch is not complete, allowing them to continue producing fetal haemoglobin.”
This insight led researchers to explore whether BCL11A could be deliberately manipulated.
The idea was that these “brakes” might be lifted to enable patients to produce more fetal haemoglobin, she says.
Manipulating gene activity
Thus, researchers began exploring how the gene could be safely targeted.
After identifying BCL11A in 2007, it took 16 years of further research to translate the discovery into a viable treatment.
“The next stage was figuring out how to manipulate BCL11A safely,” says Dr Thein.
Working independently, researchers at the Harvard Medical School in the United States, led by David G. Nathan Distinguished Professor of Paediatrics Dr Stuart H. Orkin, identified a critical region within the gene that regulates fetal haemoglobin production.
By disabling this specific region, scientists can effectively “lift the switch”, removing the suppressive effect of BCL11A without affecting its other essential roles in the body.
With the help of the revolutionary CRISPR-Cas gene editing tool, the first therapies utilising this knowledge started becoming available in 2023.
“It is a highly precise gene-editing tool that can be used to disable the specific part of BCL11A responsible for suppressing fetal haemoglobin production,” explains Dr Thein.
Increasing fetal haemoglobin benefits both beta-thalassaemia and sickle cell disease patients, although in different ways.
In beta-thalassaemia, it compensates for the lack of adult haemoglobin.
In sickle cell disease, it interferes with the sickling process that leads to complications.
However, these therapies require many resources and advanced hospital facilities.
“The current approach, known as ex vivo gene editing, is complex and demanding,” she says.
“For conditions like sickle cell disease, the entire process can take about a year and is physically very gruelling for patients.”
The treatment involves extracting the patient’s stem cells, editing them in specialised laboratories to modify the BCL11A gene, then reinfusing them into the patient after chemotherapy has cleared the bone marrow to make space for the modified cells.
“This is a vulnerable period, as patients are unable to produce sufficient blood cells and require intensive medical support,” Dr Thein explains.
Cost is another major barrier, with treatment in the US ranging from US$2mil (RM8mil) to US$3mil (RM12mil) per patient.
ALSO READ: Life-saving treatments available, but at exorbitant cost
As a result, scientists are now exploring in vivo gene editing, which delivers the gene-editing machinery directly into the body to target stem cells.
But this approach may take years before becoming widely available.
Honoured for her discovery
Dr Thein’s decades of research have not only transformed treatment for these two diseases, but have also earned global recognition.
She recently became the first Malaysian-born scientist to receive the Breakthrough Prize in Life Sciences for her discovery of BCL11A.
Currently working as the US National Heart, Lung and Blood Institute Sickle Cell Branch chief, Dr Thein shared one of the three 2026 Life Sciences prizes with Prof Orkin.
Each prize carries a cash award of US$3mil (RM12mil) – the largest for scientific achievements.
Dr Thein says that she hasn’t really thought about how she will use her share of the prize money, but may contribute some to charity, as well as her team’s research work.
ALSO READ: Dr Thein becomes first Malaysian-born scientist to win ‘Science Oscar’
The journey, she says, has not been without doubt.
“There were many moments when I had to convince myself to keep going.
“But the biology was there, I could not ignore it.”
Looking ahead, her focus remains on improving treatment access for sickle cell disease.
For alpha- and beta-thalassaemia, Dr Thein’s more recent work has already helped produce a different drug that has received approval from the US Food and Drug Administration (FDA).
“Hopefully, it will also become available in Malaysia for thalassaemia patients, and eventually be approved by the FDA for treating with sickle cell disease as well,” she says.
Already a subscriber? Log in
Get 20% OFF The Star Digital Access
Cancel anytime. Ad-free. Unlimited access with perks.


