



Ardi Izzuddin Zabidi, 16, has an ultra-rare disease that only strikes less than one in 250,000 babies.
His condition is so rare that he is the only person in Malaysia known to have it.
As his mother Maisarah Badaruddin, 46, shares, their journey started when Ardi was 16 months old.
During a regular check-up at the Klinik Kesihatan (Health Clinic), the doctor discovered that Ardi’s head was much larger than expected for his age and referred him to Tengku Ampuan Afzan Hospital in Kuantan, Pahang, where the family lives.
The doctors at the Paediatric Department there initially diagnosed him as having hydrocephalus.
Hydrocephalus occurs when there is a build-up of fluids in the ventricles within the brain, resulting in an enlarged head in babies.
However, further investigations, including a CT (computed tomography) scan, showed that this wasn’t the case.
“After two or three visits, they referred us to HKL (Kuala Lumpur Hospital),” says Ardi’s father Zabidi Ali, 53.
“They were unable to detect the problem,” Maisarah says.
It was at HKL that the doctors suspected Ardi might have a genetic condition.
A urine test known as a toluidine blue-spot test confirmed Ardi had mucopolysaccharidosis (MPS).
MPS is actually a group of inherited metabolic diseases, which are a part of a larger group of rare genetic diseases known as lysosomal storage disorders.
These disorders cause a build-up of toxic materials in the body due to the lack of certain enzymes or substances that facilitate the action of particular enzymes.
The enzymes involved in these conditions are found in the body’s lysosomes, which are the main digestive units in our cells.
In MPS, there is either a lack or improper functioning of lysosomal enzymes involved in the digestion of complex carbohydrates known as mucopolysaccharides.
Because of this, the mucopolysaccharides in the body will accumulate in the body’s cells, causing problems.
The excessive amounts of mucopolysaccharides, also known as glycosaminoglycans, excreted in the urine are what is picked up by the toluidine blue-spot test.
However, MPS has seven distinct types, as well as various subtypes.
Each distinct type is caused by a problem with one or more of the 11 lysosomal enzymes involved in the digestion of mucopolysaccharides.

Thus began a whole battery of tests to determine exactly which type of MPS Ardi had.
These included urine tests, a skin biopsy and blood tests – not only for Ardi, but also his parents – among others.
And because Malaysia did not have the capability for detailed genetic tests at that time, the blood tests had to be sent to Australia for analysis.
It was a long process, with Maisarah sharing that Ardi was finally confirmed to have MPS VII when he was three years old.
No cure, just support
At that time however, there was no cure for MPS VII.
“There was no medicine at all; they could only provide supportive and palliative care,” says Maisarah, adding that they would go to HKL every six months or a year for follow-up appointments to monitor Ardi’s progress.
As mucopolysaccharides are involved in the building of various tissues, including bone, cartilage, tendons, corneas, skin and connective tissue, MPS can affect multiple parts of the body.
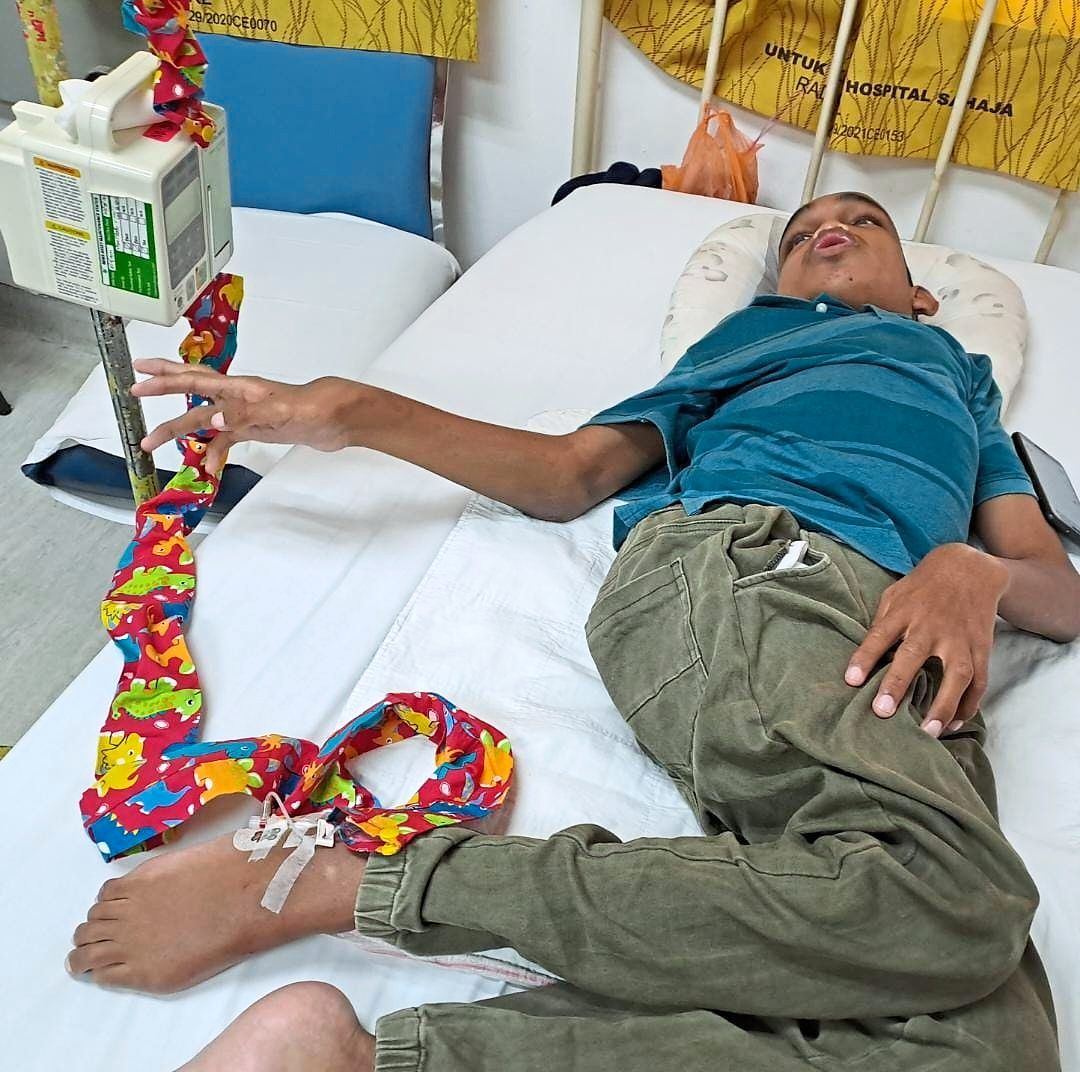
Aside from a large head (macrocephalus), some of Ardi’s symptoms include coarse facial features, short trunk dwarfism (an unusually short trunk and growth disability), cornea cloudiness, speech and hearing impairments, bone deformities, sleep apnoea, heart and lung problems, intellectual disability, and almost-constant respiratory infections.
Maisarah says that Ardi attends 10 clinics for his various symptoms, including respiratory, cardiology, eye, rehabilitation, and ear, nose and throat (ENT) clinics.
One of the sad things about MPS is that many children with it experience a period of normal development, followed by a subsequent decline in function.
Similarly, Zabidi shares that Ardi’s ability to walk and run started to deteriorate when he was around seven years old.
Maisarah adds: “He used to be able to walk and run like other kids. “But he became slower, he was smaller (compared to children his age), his hands became stiff; then at eight to nine years of age, he couldn’t walk any more, he could only sit.”
She says that he wasn’t even strong enough to push his wheelchair by himself.
However, the year that Ardi’s health started to go downhill was also the year that Zabidi and Maisarah received some news that enabled them to “see a bit of light”.
Seeing the light
In August 2014, Malaysia Lysosomal Diseases Association (MLDA) president Lee Yee Seng attended the International Symposium on Mucopolysaccharidoses and Related Diseases in Bahia, Brazil.
MLDA is a non-profit organisation that raises awareness about lysosomal storage diseases, as well as advocates for patients’ rights to a sustainable healthcare and support system.
At the symposium, Lee heard about a new drug for MPS VII, which had shown promising results in mice.
The drug called vestronidase alfa-vjbk, is a recombinant human lysosomal beta glucuronidase, which substitutes for the missing beta glucuronidase in MPS VII patients.
He managed to meet up with Dr Emil Kakkis, the CEO and president of Ultragenyx, the rare disease drug company that was developing the enzyme replacement therapy (ERT), at the event and shared with him about Ardi.
The following year in November, Zabidi, who is the vice-president of MLDA, shares that a representative from BioMarin Biotechnology Malaysia (the local branch of the US-based pharmaceutical company) reached out to him on behalf of a friend from Ultragenyx to say that they were looking for participants for the vestronidase alfa-vjbk clinical trial.
However, they would have to go through the hospital (HKL).
Dr Kakkis retired as BioMarin’s chief medical officer in 2009 before he started Ultragenyx the following year.
While at BioMarin, he had guided the development of treatments for MPS I and VI, and phenylketonuria.
The following month, Zabidi and Maisarah met up with HKL consultant paediatrician and clinical geneticist Dr Ngu Hock Lock, who would be Ardi’s healthcare sponsor for the treatment if they decided to let Ardi start it.
“Of course, we wanted it!” says Maisarah.

So Dr Ngu, who is now HKL’s Genetics Department head, began the process of bringing the drug in for Ardi.
As vestronidase alfa-vjbk is not registered in Malaysia, the hospital had to apply for a special import permit to bring it into the country.
According to Health Ministry Pharmaceutical Services senior director Norhaliza A Halim, the importation of an unregistered medicine can be applied for in the situation where it is not available in Malaysia, is needed for the treatment of a person suffering from a life-threatening illness, and there are no other suitable options or alternative treatments available.
In such cases, the healthcare facility treating the patient may submit an Import Permit application for the importation and use of an unregistered medicine that can be used to treat the patient.
She says: “This application for medicines of special approval or application to import products for the treatment of life-threatening illnesses provides a pathway to ensure the continuous access to medicines needed for the treatment of a patient in cases where there are no registered or suitable medicines or treatment options available locally.
Norhaliza, who oversees the National Pharmaceutical Regulatory Agency, adds: “The processing and evaluation of such applications will be expedited by the Pharmaceutical Services Programme in order to ensure the timely access to the medicine needed for the treatment of the patient’s condition.”
A vast improvement
Despite the availability of such processes, Zabidi shares that it still took a long while before Ardi could start his treatment because the drug was still under clinical trial and needed to go through multiple layers of approval within the ministry, up to the director-general himself.
“In March 2016, Dr Ngu called us in to explain and sign the consent form.
“We waited until November 2017 before Ardi received his first dose of the medicine,” he says.
Coincidentally, vestronidase alfa-vjbk received approval from the US Food and Drug Administration (FDA) on Nov 15, 2017.
Lee shares that Ardi totally changed, “like turning into a new leaf”, after he started receiving the treatment.
“He was so weak before the ERT, but now, every time, he will shake hands with you non-stop.
“And if he wants to push the wheelchair on his own, you cannot stop him – he’s so energetic now.”
He adds that Ardi also gets sick far less than he used to; previously, “he was so sick and had runny nose all the time, macam makan cendol (like eating cendol)”.
Zabidi agrees, saying that Ardi’s health has improved tremendously after he started the treatment.
He adds that Ardi’s intellectual abilities have also improved, e.g. he knows the cues when a trip is upcoming, like when his mother packs a bag, and he will wait for Zabidi at the door when it’s time for him to come home.
Ardi is also doing well at his special education school, Sekolah Kebangsaan Pendidikan Khas Kuantan, according to Maisarah, who says that he can follow his teacher’s instructions during lessons and has mastered new skills.
As the medicine needs to be administered intravenously (i.e. via a drip into a vein) once every two weeks over four hours, Ardi and his parents need to make the trip from the East Coast to Kuala Lumpur every fortnight.
An additional challenge is that Zabidi is based in Manjung, Perak, as a contractor for Telekom Malaysia, which means that he has to travel back to Kuantan to pick up Maisarah and Ardi before making the trip back to the West Coast.
But despite the distance and thanks to understanding bosses, they have never missed a treatment, especially since it is being given to Ardi free under Ultragenyx’s compassionate use policy.
Compassionate access
While the initial agreement was for vestronidase alfa-vjbk to be provided to Ardi as part of the drug’s clinical trial for two years (2016-2018), he is still receiving the treatment for free up to today.
At an estimated cost of about RM3.36 million a year (the dose is weight-dependent, with Ardi currently needing about 11 two-milligramme bottles each time), the price is beyond Zabidi’s and Maisarah’s salaries, especially as they also have to think about Ardi’s older sister who just started university this year.
Maisarah works as a nurse at Tengku Ampuan Afzan Hospital.
Says Dr Kakkis: “Rare diseases require a higher level of responsibility in how you handle them.
“The fact that in the whole world, we are the only company that has an MPS VII treatment... with the privilege of creating a drug like this, comes a responsibility to ensure access.
“But one of the biggest issues (for pharmaceutical companies) is the fear that if they have to treat someone, they have to treat the whole world.
“But I’ve told people that not being able to treat the whole world in some future state, is not a reason to not treat a child in front of you right now.”
Although Ultragenyx is a business (and thus, needs to make a profit), he shares that their goal is to make a profit overall, but not necessarily a profit off each and every patient.
Thus, the company’s compassionate use policy includes ensuring that qualified patients worldwide do not have to forego treatment due to financial reasons.
Such patients are evaluated on a case-by-case basis.
Patients (and doctors on behalf of their patients) can also gain access to the company’s rare disease drugs that are still under clinical trial by requesting to participate in such trials, even if they are the only one in their country.
Dr Kakkis notes that while rare disease patients can, and do, get in touch with the company (or even himself) directly to request for access to their drugs, Ultragenyx requires that a doctor be involved in the process to be medically responsible for the patient.
“While patients will request compassionate use directly from us, our response is usually to ask them to get the doctor who will be responsible for the treatment to contact us, because he is the authority we have to work through,” he says.
“There’s a particular team we have – the IST (Investigator Sponsored Trials) team – that is specifically designed to handle these cases.
“They are very knowledgeable about what to do and how to put a programme together for someone who needs to get treated.”
The doctor is considered both an “investigator” for the clinical trial and the healthcare “sponsor” for US FDA bureaucratic purposes.
Ardi’s case is the first time Ultragenyx has worked in Malaysia.
For Zabidi, Ardi’s free treatment is “macam bulan jatuh ke riba (like the moon fell on your lap)” – something that was totally unexpected.
Says Lee: “At one time, I would have said this boy is just waiting for his time, but miracles can happen.
“Ardi’s story teaches us to not give up on hope – he is my inspiration.”
Zabidi agrees, saying: “In this world, the unexpected can happen.
“If you are patient (sabar) and strong (tabah), something good may happen.”
This article is the first of a short series written as part of the US National Press Foundation’s 2022 Covering Rare Diseases: Journalism Fellowship & Global Reporting Grant. The second one will be published next Sunday (Jan 1, 2023) in StarHealth.
Already a subscriber? Log in
Get 20% OFF The Star Digital Access
Cancel anytime. Ad-free. Unlimited access with perks.


